
Our highly parallel high performance pattern processing technology is ideally suited to Next Generation Sequencing (NGS) where enormous amounts of data are produced by high throughput sequencing machines. Implemented in configurable hardware devices, such as Field Programmable Gate Arrays (FPGAs) and manycore processors, significant speed ups can be achieved over conventional software based tools. Combined with elements of our Knowledge Processing Framework (KPF) and proprietary informatics algorithms the overall effect is to enhance genomic sequence data analysis capabilities.
The KPF can be used to implement part or all of a four stage bioinformatics pipeline consisting of base calling, alignment, variant calling and filtering and annotation. In addition to if ability to significantly accelerate the entire DNA sequencing process, the KPF can:-
Increase the rate and quality of variant identification from sequence data that is generated by a variety of next generation sequencing technologies
Improve accuracy of interpretive analysis of variant data to provide novel e-diagnostic for the future and deeper understanding of disease and its application in a clinical context
Improve efficiencies in the collection, transmission, storage, archival and disposal of data used in this way.
Pipeline Overview
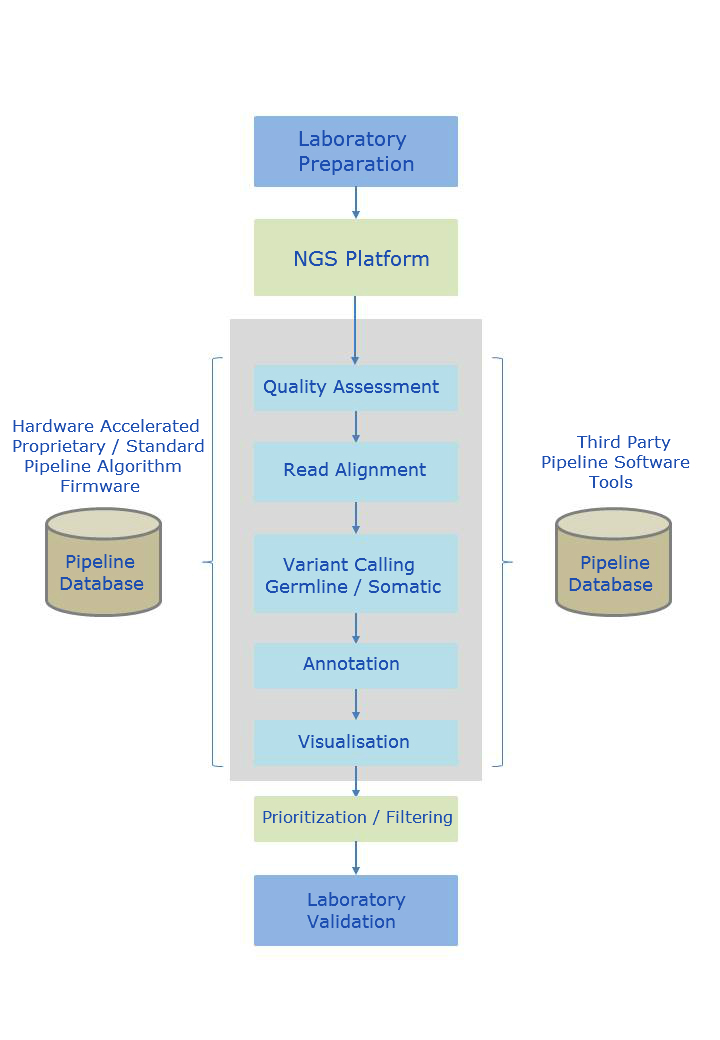
Customizable NGS pipeline configuration by mixing & matching hardware accelerated and third party software pipeline components.
The alignment process endeavours to map short nucleotide reads to a reference genome. Alignment is a non-trivial task due to the enormous number of potential start positions, unique versus non-unique mappings, and variation in base quality.
The sample genome is then compared to the reference genome where variants are identified (variant calling). The variants can take several forms such as Single Nucleotide Polymorphisms (SNPs), smaller insertions or deletions (indels), or larger structural variants of categorizations such as transversions, trans-locations and copy number variants.
The result of the latter process is a list of thousands of potential differences between the reference genome and the genome under study. Filtering and annotation are then performed to determine which of these variants are likely to contribute to the pathological process under study. Filtering involves removing variants that fit specific genetic models or are not present in normal tissue. Annotation requires looking up information about variants and identifying ones that fit the biological process.